Category
Description
Dataset Description
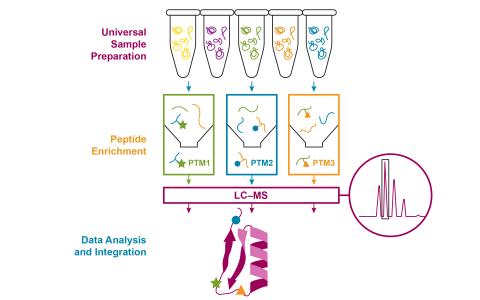
The goal of the experiment was to demonstrate that the optimized multiplexed multi-PTM profiling workflow can comprehensively and quantitatively capture dynamic changes in protein abundance, cysteine oxidation, phosphorylation, and acetylation in cytokine-induced inflammatory stress in mouse pancreatic β-cells. Global proteomic, redox proteomic, phosphoproteomic, and acetylomic were data collected from mouse Beta-TC-6 pancreatic Beta-cells, either untreated (mock) or cytokine-treated, 4, 8, and 24 hours with 4 biological replicates. Samples were digested with trypsin and Lys-C, then analyzed by LC-MS/MS. Data were searched with MS-GF+, MASIC, and MaxQuant using PNNL's DMS processing pipeline.
Data Download Reference Citation
Zhang, Tong; Gluth, Austin; Li, Xiaolu; Waters, Katrina. (2025) Integrative SP3 Workflow for Multi-PTM Proteomics Profiling (TZ-DP0). https://doi.org/10.25584/3022642
Associated Publication
Gluth A, Li X, Gritsenko MA, Gaffrey MJ, Kim DN, Lalli PM, Chu RK, Day NJ, Sagendorf TJ, Monroe ME, Feng S, Liu T, Yang B, Qian WJ, Zhang T. Integrative Multi-PTM Proteomics Reveals Dynamic Global, Redox, Phosphorylation, and Acetylation Regulation in Cytokine-Treated Pancreatic Beta Cells. Mol Cell Proteomics. 2024 Dec;23(12):100881. doi: 10.1016/j.mcpro.2024.100881. Epub 2024 Nov 15. PMID: 39550035; PMCID: PMC11700301.
Accessible Digital Data Downloads
This repository contains the following folders and files:
- TZ-DP0_SampleMetadata.xlsx: Contains sample metadata information including descriptors, experimental conditions, cell lines (if applicable)
- TZ-DP0_ExpressionData.xlsx: Contains the relative protein abundances; and relative cysteine thiol oxidation, phosphorylation, and acetylation levels of Beta-TC-6 pancreatic cells. Fold changes and statistics are also provided.
Total Download Size: 20.5 MB, Zipped
Linked Primary Data
Primary liquid chromatography-mass spectrometry (LC-MS) raw measurement data are openly accessible for download at the Mass Spectrometry Interactive Virtual Environment (MassIVE) community repository under the accession MSV000095264.
Funding Acknowledgments
The research data described here was funded in whole or in part by the Predictive Phenomics Initiative (PPI) at Pacific Northwest National Laboratory (PNNL). This work was conducted under the Laboratory Directed Research and Development Program at PNNL. A portion of this research was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy (DOE) Office of Science located at PNNL. PNNL is a multiprogram national laboratory operated by Battelle for the DOE under Contract No. DE-AC05-76RL01830.
Citation Policy
In efforts to enable discovery, reproducibility, and reuse of PPI-funded project dataset citations in accordance with best practices (as outlined by the FORCE11 Data Citation Principles), we ask that all reuse of project data and metadata download materials acknowledge all primary and secondary dataset citations and corresponding journal articles where applicable.
Data Licensing
Creative Commons Attribution 4.0 International (CC BY 4.0)
Projects (2)
People (1)
